

Expert analysis at the intersection of AI, bioinformatics, and genomics. Breaking down the science shaping precision medicine — from whole-genome sequencing and liquid biopsy to spatial transcriptomics, proteomics, and metabolomics — grounded in peer-reviewed literature and written for the people building the next generation of life sciences products.
From Plasma to Pixels: How AI Is Unifying Liquid Biopsy and Spatial Transcriptomics for Precision Oncology
Liquid biopsy tells you what a tumor is shedding into the bloodstream — circulating DNA fragments, methylation patterns, copy number shifts. Spatial transcriptomics tells you where resistance lives in tissue — which cell subpopulations cluster near vasculature, where immune exclusion is occurring, which signaling pathways are active at the tumor margin. Until recently, these two fields evolved in parallel, with separate platforms and separate toolkits. AI is beginning to bridge them. Foundation models, multimodal fusion architectures, and deep learning pipelines are connecting plasma-level signals to tissue-level maps. The convergence is reshaping how we detect, monitor, and treat cancer.

Multi-Omics Integration — Why One Data Type Is Never Enough
Biology doesn't operate one layer at a time. A single mutation can rewire transcription, reshape the proteome, and redirect metabolic flux — yet most studies still capture only one data type. Multi-omics integration combines genomics, transcriptomics, proteomics, and metabolomics to reveal what no single platform can see alone. From TCGA's colorectal cancer breakthroughs to tools like MOFA+ and DIABLO, this article explains why integrated analysis is now the clinical standard — and how to design studies that get it right from the start.

CAP/CLIA Compliance in Bioinformatics Pipelines
Clinical bioinformatics pipelines aren't just a science problem — they're a regulatory one. CAP and CLIA require validation, version control, and documented SOPs for every computational step. This article breaks down what compliance actually demands, where most labs fall short, and how to close the gaps.

From Sequencing Data to Clinical Insight
Sequencing is only the first step. The real work happens in the bioinformatics pipeline — the computational engine that transforms raw FASTQ files into somatic variants, expression profiles, and clinical insights. This article breaks down every stage, from QC to reporting, in plain language.
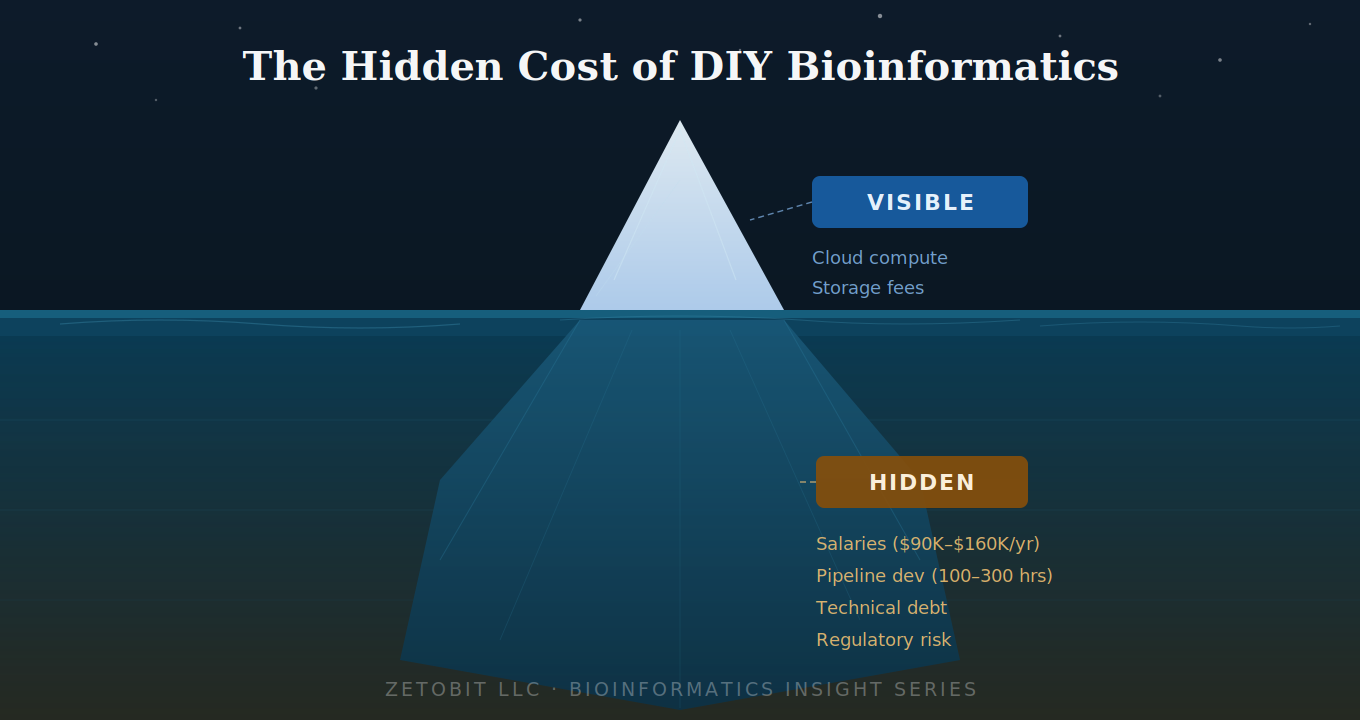
The Hidden Cost of DIY Bioinformatics
Building bioinformatics in-house looks affordable — until you account for what's below the waterline. Salaries, pipeline development, technical debt, and regulatory gaps routinely 3–5× the visible compute costs. For early-stage biotechs and clinical programs, outsourcing to a validated partner is almost always the smarter financial and strategic decision.

Bulk RNA-seq vs. scRNA-seq — When to Use Each (and When You Need Both)
Not all transcriptomic experiments are created equal. Bulk RNA-seq delivers statistical power across large cohorts; scRNA-seq reveals the hidden cellular heterogeneity bulk data averages away. Choosing the wrong method costs months. This guide helps you match your biological question to the right technology — or decide when you need both.
Liquid Biopsy Intelligence: ctDNA in the Age of AI
What if detecting cancer — or monitoring its response to treatment — required nothing more than a blood draw?
That question is no longer hypothetical. Circulating tumor DNA (ctDNA) analysis has transformed oncology by enabling a non-invasive window into cancer biology that tissue biopsy simply cannot match. Tumors continuously shed small fragments of DNA into the bloodstream during cell death. These fragments — typically just 160–200 base pairs long and comprising as little as 0.01% of total circulating DNA — carry the full mutational and epigenetic fingerprint of the originating tumor. Capturing and interpreting that signal is the promise of liquid biopsy.
Five Clinical Frontiers
The clinical applications of ctDNA are broad and rapidly maturing. Multi-cancer early detection (MCED) platforms now screen for more than 50 cancer types from a single plasma sample using genome-wide methylation profiling — the NHS-Galleri trial alone enrolled 140,000 participants. After curative surgery, ctDNA-based molecular residual disease (MRD) monitoring predicts relapse with striking precision: in colorectal cancer, ctDNA positivity post-resection carried a hazard ratio of nearly 12 for inferior disease-free survival. For patients with advanced gastrointestinal cancers, the GOZILA study demonstrated that ctDNA-guided therapy matching significantly improved overall survival compared to unmatched patients. And for hematologic malignancies like multiple myeloma, serial ctDNA sequencing post-CAR T-cell therapy now tracks both tumor burden and CAR-T cell kinetics non-invasively — in real time.
Where AI Changes Everything
The ctDNA signal space is extraordinarily rich: mutations, DNA methylation, copy number variations, fragment size distributions, end-motif frequencies, nucleosome positioning. No single conventional tool can integrate all of it. That's where artificial intelligence steps in.
Deep learning frameworks like MRD-EDGE achieve approximately a 300-fold improvement in signal-to-noise ratio over conventional WGS-based MRD detection — enabling tumor monitoring at ctDNA fractions that were previously undetectable. Multi-modal platforms like SPOT-MAS simultaneously analyze methylomics, fragmentomics, CNV, and end motifs from a single low-depth sequencing run. AI tools like MetaCH now distinguish true tumor variants from clonal hematopoiesis — a major source of false positives — without requiring matched white blood cell sequencing. And interpretable ML models like PRIME provide SHAP-based per-patient explanations that oncologists can actually act on.
The field is converging on fourth-generation architectures that jointly model all cfDNA feature types, achieving 72.4% sensitivity at 97% specificity across multiple cancer types — performance no single modality can approach alone.
The Bottom Line
ctDNA liquid biopsy, powered by AI, is not a future technology. It is being validated in trials of hundreds of thousands of patients, earning FDA approvals, and reshaping how we detect, monitor, and treat cancer. At Zetobit, we help life sciences organizations navigate this landscape — from pipeline design to multi-modal bioinformatics analysis.

AI Applications in ctDNA Analysis and Liquid Biopsy
Every time a tumor grows, it leaves behind evidence — tiny fragments of mutated DNA shed into the bloodstream. Circulating tumor DNA (ctDNA) represents one of the most promising frontiers in cancer diagnostics, offering a non-invasive window into tumor biology that tissue biopsies simply cannot match.
Through a routine blood draw, ctDNA analysis can detect cancer before symptoms appear, track residual disease after surgery, monitor treatment response in real time, and reveal the emergence of resistance mutations — all without a scalpel. This is the promise of liquid biopsy.
The GALAXY study (n=2,240 colorectal cancer patients) demonstrated that ctDNA positivity after curative resection predicted inferior disease-free survival with a hazard ratio of 11.99 — among the most powerful prognostic biomarkers ever reported in solid tumor oncology.
But ctDNA is extraordinarily rare in blood — often comprising less than 0.1% of total cell-free DNA — and its signals are buried beneath sequencing noise, normal cell DNA, and confounding variants from age-related clonal hematopoiesis. This is precisely where artificial intelligence changes the game.
In this review, we survey 20 landmark publications at the intersection of AI and ctDNA, tracing the field's evolution from conventional variant callers (MuTect2, CAPP-Seq, ddPCR) to a new generation of deep learning frameworks, ensemble classifiers, and multimodal integration platforms. Tools like MRD-EDGE achieve a 300-fold improvement in signal-to-noise ratio over prior WGS methods; MetaCH eliminates the need for matched white blood cell sequencing by learning to distinguish clonal hematopoiesis from true tumor variants; and PRIME integrates ctDNA-MRD with clinical features using interpretable ML to predict NSCLC progression — validated across six independent cohorts.
At the population scale, MCED platforms like Galleri (GRAIL) deploy deep learning across genome-wide methylation profiles to simultaneously screen for 50+ cancer types. The NHS-Galleri trial enrolled 140,000 participants — a pivotal moment in AI-enabled cancer screening. Closer to the clinic, FDA-approved Guardant Shield achieves 83% sensitivity for colorectal cancer detection from a single blood draw.
The full review covers conventional tools vs. AI architectures, landmark platforms, disease-specific applications across NSCLC, CRC, hematologic malignancies, and GI cancers — and the road ahead, from fourth-generation multimodal AI to foundation models for cfDNA.

Building for the Long Run: Reproducibility in Bioinformatics Pipelines
A genomics pipeline that produces different results on two different machines—or even on the same machine six months later—is not a reliable scientific instrument. Yet this is a surprisingly common reality in the field. Software versions drift, operating system libraries are updated, and environments are configured differently across institutions.
For organizations operating under CAP/CLIA compliance, regulatory submissions, or multi-site collaborative studies, pipeline reproducibility is not a luxury—it is a foundational requirement. The bioinformatics community has responded with a mature ecosystem of tools built specifically to solve this problem.
In this article, we cover:
How Docker and Singularity / Apptainer establish a portable, immutable execution environment for genomic workflows
The leading workflow management systems—Nextflow, Snakemake, Cromwell / WDL, and CWL—and how each fits different organizational needs
Constellation, a clinical-grade containerized pipeline framework co-authored by the Zetobit team, and its deployment at the University of Kentucky Medical Center
Complementary tools—Conda, Git, DVC, and cloud platforms—that complete the reproducibility stack
Whether you are building clinical diagnostic pipelines or multi-omics research workflows, this guide maps the full reproducibility stack and the practical decisions that come with it.

AI Is Transforming Bioinformatics. Bioinformaticians Are Still Irreplaceable
A landmark 2025 survey in Briefings in Bioinformatics reviewed AI applications across two decades of publications and documented milestones that were unthinkable a decade ago: protein structure prediction at near-atomic accuracy, protein design success rates up to 92%, and cancer detection AUC approaching 0.93. Foundation models now perform zero-shot cell-type annotation. Deep learning variant callers outperform classical statistical methods on both short- and long-read data.
The pace of progress is extraordinary. But the question that matters most for the life sciences industry is not "can AI do this?" — it is "who ensures the output is scientifically credible, biologically meaningful, and safe to act on?"
That answer has not changed.
The 2024–2025 literature identifies five domains where trained human judgment remains irreplaceable:
Study design and QC. No AI model chose the sequencing depth, selected the patient cohort, or determined whether batch effects represent biological signal or technical artifact. That judgment requires experimental context no model possesses.
Multi-omics integration. LLMs and deep learning models struggle with the sparsity, noise, and cross-layer biology required to integrate genomic, transcriptomic, proteomic, and metabolomic data meaningfully across heterogeneous systems.
Data quality and curation. In AI-accelerated pipelines, uncurated data problems are amplified, not filtered out. Bioinformaticians are the primary gatekeepers — a role that cannot be automated because it requires biological plausibility judgments grounded in domain expertise.
Regulatory and clinical contexts. In CAP/CLIA-regulated environments and FDA submissions, every analytical decision must be documented, defensible, and auditable. AI black-box models do not currently satisfy those requirements. Human sign-off is non-negotiable.
Novel problem framing. AI excels in well-characterized problem spaces with abundant training data. For rare diseases, understudied organisms, and emergent pathogens, bioinformaticians with deep domain expertise are required to ask the right questions and identify when a model is extrapolating dangerously outside its training distribution.
The market for generic bioinformatics — standard pipelines, standard outputs — is being commoditized. The market for expert interpretation, custom multi-omics integration, regulatory-grade analysis, and novel problem solving is growing.
The positioning opportunity is not to compete with AI tools. It is to be the expert layer that makes AI outputs scientifically credible and actionable for clients who lack that expertise in-house.

Beyond the Genome: Integrating WGS and RNA-seq for Clinical Diagnosis
Whole-genome sequencing has given clinicians an unprecedented view of the human genome — but sequence alone doesn't always tell the full story. A variant sitting in a splice region may look suspicious on a DNA readout and go no further, classified as a variant of uncertain significance (VUS) and filed away while a patient's diagnosis stalls. RNA sequencing changes that equation entirely.
By capturing the transcriptional output of the genome — which genes are switched on, how their transcripts are spliced, where expression has gone off-script — RNA-seq transforms ambiguous DNA findings into actionable clinical evidence. When a deep intronic variant quietly recruits a cryptic exon, RNA-seq catches the aberrant transcript. What was a VUS becomes likely pathogenic. What was an impasse becomes a diagnosis.
This is the promise of integrated WGS + RNA-seq: not two parallel tests, but a single interpretive framework in which genomic and transcriptomic data are read together. Studies in rare disease have shown diagnostic uplift of 7–35% when RNA-seq is layered onto genome or exome sequencing — a meaningful gain for patients who have often already exhausted conventional workups.
In oncology, the case is equally compelling. Fusion transcripts like BCR::ABL1 and EML4::ALK — critical drivers of therapeutic decision-making — are best detected at the transcript level. RNA-seq simultaneously reveals the expression landscape, identifies splice isoforms that underlie resistance, and, when combined with WGS mutational signatures, enables a more complete tumor portrait than either modality alone.
The computational infrastructure is maturing to match. Tools like OUTRIDER and FRASER operationalize aberrant expression and splicing detection against reference cohorts. SpliceAI links genomic variants to their transcript-level consequences. And the outputs slot directly into ACMG/AMP classification frameworks, enabling defensible, audit-ready clinical reporting.
Challenges remain — RNA quality requirements, tissue-type limitations for Mendelian disease, and evolving reimbursement pathways among them. But the trajectory is clear. As CAP/CLIA-validated RNA-seq panels proliferate and long-read technologies begin to unify genomic and transcriptomic capture in a single assay, integrated sequencing is moving from research innovation to clinical standard.
The genome tells you what a patient has. The transcriptome tells you what it's doing. Used together, they finally give clinicians the full picture.

Multimodal AI Analysis of Genomics Data
For decades, bulk RNA sequencing gave us a powerful but incomplete picture — the average voice of thousands of cells speaking at once. Then single-cell RNA-seq gave each cell its own microphone. Now, spatial transcriptomics has placed those cells back on a map, restoring the tissue geography that dissociation methods destroy.
Three revolutionary technologies. Three incomplete pictures. One transformative opportunity.
The frontier of computational biology is no longer about extracting more from any single modality — it's about teaching AI to see all three at once.
In our latest literature review, we synthesized 147 curated publications spanning bulk RNA-seq, scRNA-seq, spatial transcriptomics, and the AI frameworks unifying them. What emerges is a field in rapid convergence. Foundation models pretrained on tens of millions of cells — including scGPT, scFoundation, and Nicheformer — are now capable of zero-shot prediction across tasks that didn't exist when they were trained. Tools like Tangram and Cell2location map single-cell identities onto spatial coordinates with striking accuracy. And models like SpaRx are already predicting spatially heterogeneous drug resistance within individual tumors, identifying which cell ecosystems to target — before a patient receives a therapy that won't work for them.
The clinical implications are immediate and substantial. Existing bulk RNA-seq datasets — millions of samples already in repositories like GEO — contain far more biological intelligence than differential expression analysis alone recovers. AI deconvolution and transfer learning are unlocking that latent signal now. At the same time, spatial foundation models represent the leading edge of what's coming: tissue-scale maps of immune activation, drug resistance, and cellular crosstalk, resolved at the level of individual cells.
This is the era when genomics data stops being an output and starts being a language — one that AI is learning to read, speak, and reason in.

AI and Multi-modal AI glossary
If you've picked up a genomics AI paper in the last two years, you've almost certainly run into terms like foundation model, variational autoencoder, contrastive learning, or spatial domain identification — often in the same abstract, often without explanation.
That's the problem this guide is designed to solve.
At Zetobit, we spend a lot of time at the intersection of cutting-edge AI and applied bioinformatics. And one thing we've noticed consistently: the barrier to understanding multimodal AI in genomics isn't the science itself — it's the vocabulary. Once you understand what the terms actually mean, the logic of the field clicks into place quickly.
So we distilled our 147-publication literature review into a plain-language glossary of the 19 terms that matter most.
Here's what's inside:
The guide is organized into four sections. We start with the three core data modalities — Bulk RNA-seq, single-cell RNA-seq, and Spatial Transcriptomics — and explain what each one captures, what it misses, and why no single modality tells the whole story. We then walk through five types of AI models used to analyze these data: foundation models, variational autoencoders, graph neural networks, transformers, and contrastive learning approaches. Each gets a plain-English definition, a full explanation of how it works, and an everyday analogy to make it stick.
The final two sections cover how data integration actually works in practice — batch correction, latent spaces, deconvolution, transfer learning — and the biological tasks these tools are built to solve, from mapping the tumor microenvironment to predicting drug resistance at single-cell resolution.
Whether you're a bench scientist trying to evaluate an AI platform, a biotech executive reading a vendor white paper, or a clinician wondering what "spatial foundation model" means in the context of your patients — this guide was written for you.
No PhD required.
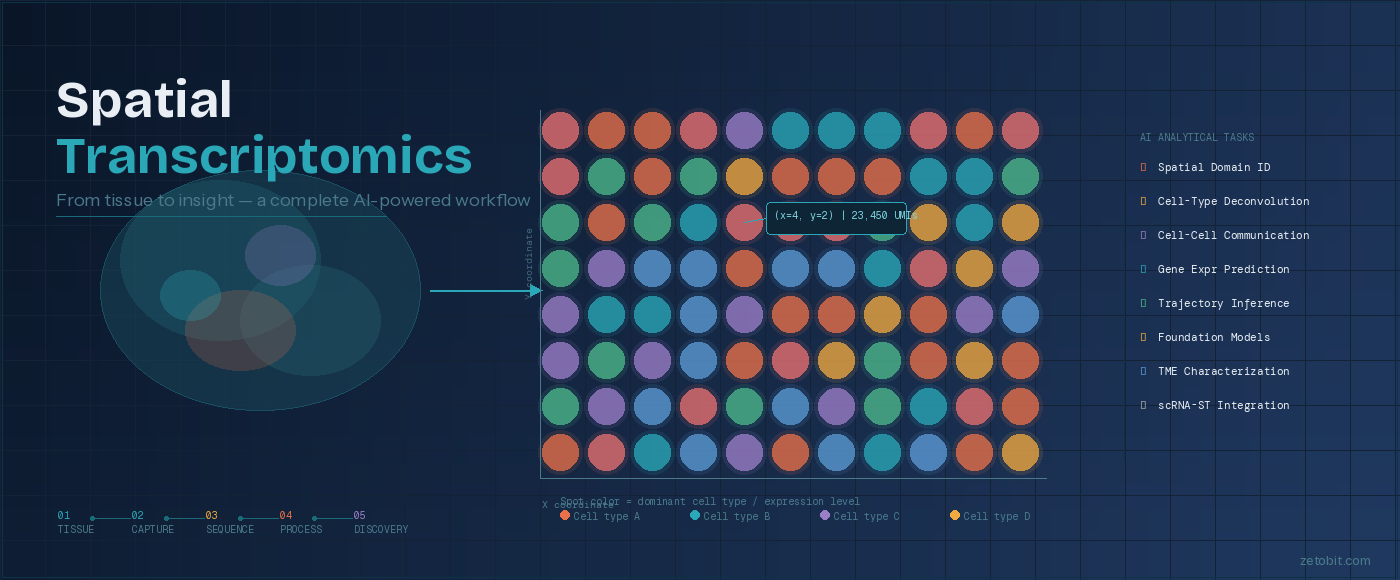
Spatial Transcriptomics: From Tissue to Insight
How AI is transforming the way we decode biology — one spot at a time
For decades, genomics told us what genes a cell expressed — but never where. Spatial transcriptomics changes that equation entirely. By combining sequencing or imaging technologies with precise tissue coordinates, researchers can now generate a gene expression map of intact tissue, revealing how biology is organized in space, not just in sequence.
The workflow begins with a tissue section — fresh-frozen or FFPE — placed on a barcoded slide. As mRNA molecules bind to spatial probes, each transcript is tagged with both its gene identity and its XY location in the tissue. That data is then processed through next-generation sequencing or fluorescence imaging, producing a spatial count matrix: a full gene expression profile for every spot across the tissue section.
What happens next is where the field has transformed dramatically. A growing ecosystem of AI and machine learning tools now extracts biological meaning from these maps — identifying tissue domains, deconvolving mixed cell populations within individual spots, modeling how cells communicate across space, and even predicting gene expression directly from histology images. Zetobit's review of this landscape covers 48+ tools spanning eight analytical categories, from spatial domain identification (STAGATE, GraphST) to foundation models purpose-built for spatial biology (Nicheformer, HEIST).
Platform choice matters too. Sequencing-based technologies like 10x Visium (~55 µm resolution, ~5,000 genes) offer broad transcriptomic coverage, while imaging-based platforms like MERFISH and CosMx push resolution to the subcellular level with targeted gene panels. The right choice depends on the biological question — and increasingly, the answer involves combining multiple modalities.
Spatial transcriptomics is no longer an experimental novelty. It is becoming a standard layer of the multi-omic stack — one that Zetobit is helping life science organizations navigate from platform selection through AI-powered discovery.

From Raw Signal to Clinical Report: How AI is Transforming Whole Genome Sequencing Analysis
FROM RAW SIGNAL TO CLINICAL REPORT:
HOW AI IS TRANSFORMING WHOLE GENOME SEQUENCING ANALYSIS
Zetobit · Bioinformatics Insight Series
────────────────────────────────────────────────────────
A whole human genome contains over three billion base pairs. Sequencing it now takes hours and costs less than $200. What still takes time — sometimes weeks — is making sense of the result: identifying which of the millions of variants in that genome are clinically meaningful, what they imply for a patient's health, and what a clinician should do next. Artificial intelligence is compressing that timeline dramatically, without removing the expert judgment that makes the results trustworthy.
Modern whole genome sequencing (WGS) analysis unfolds in five distinct layers, each one transforming raw data into progressively more actionable information.
It begins with the raw signal from the sequencer itself — billions of electrical pulses or fluorescence traces representing the passage of DNA through a detection chamber. Deep learning models now decode those signals into readable sequence with per-read accuracy above 99%, a level that was unachievable just five years ago.
From there, aligned reads are compared against a reference genome to identify variants — the positions where an individual's DNA differs from the population standard. Tools like DeepVariant, built on convolutional neural networks, have significantly reduced false positive rates compared to classical statistical callers, particularly for insertions, deletions, and structurally complex regions. A typical WGS run identifies four to five million such variants.
The harder challenge is interpretation: determining which of those millions of variants actually matter. AlphaMissense, DeepMind's pathogenicity prediction model, classifies 71 million possible missense mutations across the human proteome with agreement rates that substantially exceed prior computational tools. AI-assisted variant prioritization is shortening the time to diagnosis in rare disease programs from months to days.
The final analytical layer is clinical reporting — translating a prioritized variant list into a document a physician can act on. Large language models can now draft substantial portions of these reports automatically, reducing turnaround time by 60% or more in high-volume settings. The draft still requires expert review and sign-off. The AI accelerates the process; it does not replace the judgment.
That judgment — from bioinformaticians who design and validate the pipeline, from clinical scientists who interpret results in context, from the regulatory frameworks that govern genomic medicine — remains the indispensable layer above and around all five analytical stages. The role of the expert is not shrinking; it is concentrating in higher-value work.
Whole genome sequencing is moving toward becoming a routine clinical instrument. The analytical infrastructure required to make that transition safely is being built now, with AI embedded at every layer. The organizations defining precision medicine's next chapter are those investing in AI-literate genomics capability — paired with the expert partnerships to deploy it responsibly.
────────────────────────────────────────────────────────
Zetobit, LLC · Bioinformatics Consulting · Lexington, KY

Mapping the Invisible: How Three Generations of RNA Sequencing — and Artificial Intelligence — Are Revealing the Hidden Geography of Life
For three decades, scientists have asked the same fundamental question: which genes are active, in which cells, and why? The answer has evolved dramatically — from measuring blended averages across millions of cells, to resolving individual cell identities one at a time, to the current frontier: capturing gene expression at precise spatial coordinates within an intact, living tissue architecture.

Spatial Transcriptomics and Artificial Intelligence: A Comprehensive Literature Review
For decades, understanding how genes behave across tissues meant homogenizing them first — grinding biology into an averaged signal and hoping the interesting parts survived. That era is ending.
Spatial transcriptomics preserves what dissociation-based methods destroy: the precise location of every cell within intact tissue architecture. Combine that spatial fidelity with artificial intelligence, and the result is a fundamentally new way to interrogate disease — one where where a cell lives is inseparable from what it does.
In this comprehensive literature review, Zetobit's Bioinformatics Insight Series surveys 48 primary publications on AI-powered spatial transcriptomics, drawing on a curated corpus that spans the full arc from core deep learning methods to disease-specific clinical applications.
What we cover:
The review traces the technological evolution from bulk RNA-seq through single-cell RNA-seq to spatially resolved transcriptomics, then dives into the AI architectures making it actionable. Graph neural networks (STAGATE, GraphST) have emerged as the dominant paradigm for modeling tissue as a spatial graph, achieving state-of-the-art domain identification in tissues from brain cortex to colorectal tumors. Variational autoencoders underpin cell-type deconvolution tools like cell2location and DestVI, resolving the cellular mixtures hidden within each sequencing spot. And transformer-based models — including BLEEP and GHIST — are now predicting transcriptome-wide gene expression directly from routine H&E histology images, at a fraction of the cost of sequencing.
The clinical implications are substantial. In oncology, AI-powered spatial analysis is mapping tumor microenvironment architecture with enough precision to predict immunotherapy response, characterize drug resistance niches, and identify spatially restricted cell-cell signaling axes as combination therapy targets. In neuroscience, spatial transcriptomics has produced the first in situ molecular maps of Alzheimer's plaque-proximal gene expression, Parkinson's neuroinflammatory microenvironments, and laminar-specific schizophrenia transcriptional signatures. Across liver, kidney, heart, placenta, and infectious disease, the spatial dimension is revealing pathogenic mechanisms that bulk and single-cell data fundamentally cannot see.
The newest frontier: spatial foundation models. Nicheformer, trained on 110 million cells across 73 tissue types, enables zero-shot cell annotation and niche prediction across diseases and platforms never seen during training. These models mark a qualitative shift — from task-specific tools to general-purpose spatial AI capable of transferring biological knowledge across the entire breadth of human tissue biology.
We also address the field's open challenges honestly: the resolution-coverage tradeoff that no platform has yet fully solved, the multimodal integration problem, and the regulatory and data standardization barriers that stand between today's research tools and tomorrow's clinical diagnostics.
The pace of innovation in 2024–2025 alone — with landmark tools and benchmarks published in Nature Methods, Nature Communications, Cancer Cell, and Briefings in Bioinformatics — signals a field on the cusp of broad clinical adoption. We are not at the middle of the spatial transcriptomics revolution. We are at its beginning.

6 Big Ideas from the Frontier of AI in Genomics
AI is changing how we read, interpret, and act on genomic data. Here are six ideas — explained simply — drawn from our literature review series on AI in DNA sequencing and gene expression analysis.
THE 6 CONCEPTS
01 — Reading DNA Faster and More Accurately WGS · Variant Calling
AI tools like DeepVariant scan billions of DNA letters and flag mutations the way photo software spots faces — quickly and with far fewer errors than older methods.
02 — Finding the Variants That Actually Matter WGS · Variant Interpretation
Your genome differs from a reference at millions of spots. AI models like AlphaMissense sift through those differences and score which ones are likely to cause disease — saving scientists weeks of manual review.
03 — Hearing Which Genes Are Active RNA-seq · Transcriptomics
RNA sequencing captures which genes are switched on in a tissue at a given moment. AI cuts through the noise to surface the signals that matter — critical for drug development and biomarker discovery.
04 — AI That Learned the Language of DNA Foundation Models · LLMs
Just as ChatGPT learned from text, genomic AI models learn from DNA sequences. Once trained, they can be applied to entirely new biological problems — with minimal extra data needed.
05 — Automated Clinical Reports WGS · Clinical AI
Writing a genomic report used to take days of expert work. AI can now draft one in hours — pulling together findings, flagging uncertainties, and leaving the final call to the clinician.
06 — AI Computes. Experts Decide. Strategy · Human Judgment
AI is a powerful tool, not a replacement. Bioinformaticians design the experiments, validate the outputs, and are accountable for results that affect patients and drug programs. That judgment cannot be automated.

Applications of AI in scRNAseq analysis
Every cell in your body carries the same DNA — yet a brain cell, an immune cell, and a pancreatic cell look and behave completely differently. Understanding why requires looking at individual cells one at a time. That's exactly what single-cell RNA sequencing (scRNA-seq) does.
Traditional RNA sequencing blends signals from millions of cells together, giving you an average. It's like trying to understand a crowd by listening to everyone talk at once. scRNA-seq listens to each person individually — revealing rare cell types, hidden disease signals, and biological changes that would otherwise go undetected.
The catch? A single experiment can generate data on millions of cells across tens of thousands of genes. That's an enormous amount of information — too much for conventional analysis tools to handle well. This is where artificial intelligence comes in.
In our latest review, we walk through how AI is transforming scRNA-seq analysis — covering 48 key studies across a range of methods and disease areas. AI tools now automate cell identification, predict how cells respond to drugs or genetic changes, map how cells communicate with each other, and reconstruct the step-by-step processes of development and disease.
The real-world impact is already showing. One AI model identified new drug targets for heart disease using data from just a handful of patients. Another can predict the effects of genetic changes before any lab experiment is run — dramatically speeding up drug discovery.
We cover applications across cancer, infectious disease, neuroscience, and precision medicine — and look ahead to where the field is going next.
Single-cell biology is generating the data. AI is learning to read it.

Applications of AI in Bulk RNAseq
Bulk RNA sequencing has quietly become one of the most powerful proving grounds for artificial intelligence in biomedicine. The technology, which quantifies the averaged transcriptional output of millions of cells in a single assay, now underpins more than 60% of all next-generation sequencing projects worldwide — and the public archives it has generated, including over 6.97 million biosamples in NCBI's Gene Expression Omnibus, have created a data substrate of extraordinary depth.
Our structured review of 32 publications maps the AI landscape across six thematic domains: foundation models, cancer classification and prognosis, drug response prediction, tumour microenvironment deconvolution, data augmentation via generative modelling, and multimodal integration. The centrepiece is BulkFormer, a 150-million-parameter transformer pre-trained on over 500,000 human transcriptomic profiles — the first foundation model designed explicitly for bulk, not single-cell, data. Paired with BERT-style approaches like BulkRNABert, these models are redefining what pan-cancer classification and survival prognosis can look like at scale.
Beyond the headline models, the review surfaces several under-discussed tensions: the gap between pseudo-bulk benchmarking and authentic bulk validation; the challenge of interpretability as transformer architectures grow more opaque; and the surprisingly competitive performance of simple baselines against complex deep learning for survival prediction. The path to clinical translation — through regulatory frameworks, bias mitigation, and reproducible benchmarking — demands as much attention as architectural innovation.