
Mapping the Invisible: How Three Generations of RNA Sequencing — and Artificial Intelligence — Are Revealing the Hidden Geography of Life
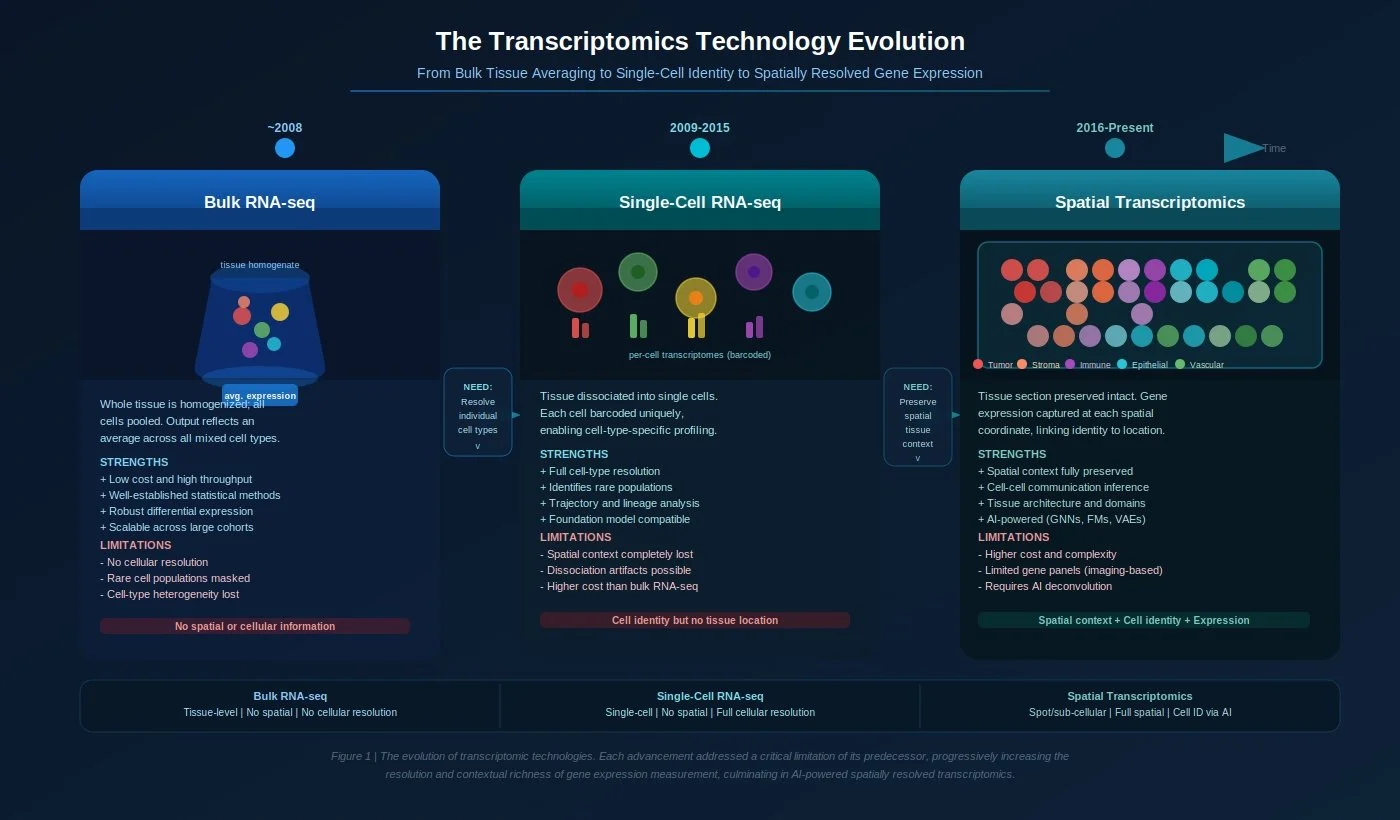
Bulk RNA-seq — The Foundation (Pre-2009 to ~2015)
Whole tissue samples are homogenized and sequenced together, producing a single averaged gene expression profile for millions of mixed cells simultaneously.
Think of it like blending an entire fruit salad into a smoothie and then trying to identify each individual fruit from the flavor — the signal is real, but the individual components are lost.
Extremely powerful for population-level comparisons: identifying which genes are turned on or off between healthy and diseased patient cohorts, classifying cancer subtypes, and discovering disease biomarkers across large sample collections.
Statistical tools like DESeq2 and edgeR established rigorous frameworks for differential gene expression, underpinning thousands of studies in cancer biology, immunology, and drug development.
AI extended bulk RNA-seq further still — foundation models trained on bulk transcriptomes can now predict drug response, deconvolute approximate cell-type proportions from mixed signals, and classify molecular subtypes with high accuracy.
Core limitation: Every measurement is an average. A tumor sample that is 30% immune cells, 50% cancer cells, and 20% stromal cells produces one blended number per gene — making it impossible to know which cell type is driving any given signal.
Single-Cell RNA-seq — The Resolution Revolution (~2009–2016)
Microfluidic droplet platforms (10x Genomics, Drop-seq) enabled sequencing of the complete transcriptome of thousands to millions of individual cells in a single experiment, each with a unique molecular barcode.
Think of it like separating the fruit salad before analysis — now every strawberry, grape, and mango can be profiled independently.
The impact was transformative: the Human Cell Atlas project used scRNA-seq to systematically map every major human organ at cellular resolution. In oncology, it revealed extraordinary intratumoral heterogeneity — drug-resistant subclones, rare cancer stem cell populations, and the complex immune ecosystems within tumors that could never be seen in bulk data.
AI tools — including transformer-based foundation models like scGPT and Geneformer, trained on tens of millions of single-cell profiles — can now generalize across tissues, diseases, and platforms, enabling zero-shot cell-type annotation and biological inference at unprecedented scale.
Core limitation: Profiling individual cells requires breaking apart the tissue first — a process called dissociation. This irreversibly destroys the spatial relationships between cells. A T cell identified in a dissociated tumor sample cannot be traced back to whether it was infiltrating the tumor core, sitting at the invasive margin, or clustering near a blood vessel. An oligodendrocyte cannot be linked to the cortical layer it came from. Where a cell lives is often inseparable from what it does — and scRNA-seq cannot answer that question.
Spatial Transcriptomics — Expression in Context (2016–Present)
Spatial transcriptomics captures gene expression directly from intact, undissociated tissue sections. A tissue slice is placed onto a capture array printed with spatially indexed barcodes; RNA molecules from overlying cells bind to these barcodes and are sequenced, preserving both the expression profile and the precise X-Y coordinates of every transcript within the tissue.
Think of it like taking a high-resolution photograph of the fruit salad before touching it — every piece is identified, characterized, and mapped to its exact position on the plate.
The foundational technology was established by Ståhl et al. in their landmark 2016 Science paper, named one of the Methods of the Year. It spawned a rapidly expanding ecosystem of platforms spanning a resolution-coverage spectrum: sequencing-based platforms (10x Visium, Slide-seq, Stereo-seq) deliver transcriptome-wide coverage at 10–55 µm spot resolution, while imaging-based platforms (MERFISH, seqFISH+, CosMx, Xenium) achieve single-molecule and sub-cellular resolution for targeted gene panels of up to 10,000 genes.
What AI unlocks in spatial data:
Spatial domain identification — Graph neural networks (STAGATE, GraphST) group spots into coherent anatomical tissue regions by jointly modelling gene expression and spatial proximity, recovering cortical layers, tumor boundaries, and immune niches with near-histological precision.
Cell-type deconvolution — Bayesian and deep learning models (cell2location, Tangram) decompose the mixed cellular signals within each sequencing spot into their constituent cell-type proportions, effectively achieving single-cell resolution from lower-resolution data.
Cell-cell communication — Spatial CCC tools (COMMOT, CellChat v2) identify which ligand-receptor signalling pairs are active between physically co-localised cell populations, producing spatially explicit wiring diagrams of tissue communication that are impossible from dissociated data.
Gene expression prediction from histology — Transformer models (GHIST, BLEEP) learn to predict transcriptome-wide gene expression patterns directly from H&E stained images, extending spatial expression inference to vast archives of existing clinical pathology slides without any new sequencing.
Foundation models — Spatially aware models like Nicheformer — trained on 110 million cells spanning 73 tissue types — enable zero-shot transfer of spatial biological knowledge across any tissue, disease, or platform.
Why this matters clinically: The tumor microenvironment, the spatial organisation of immune infiltrates, the zonation of metabolic activity in liver disease, the laminar distribution of neurodegeneration in Alzheimer's — these are all spatial phenomena that bulk and single-cell sequencing can only approximate. Spatial transcriptomics, powered by AI, maps them directly, providing a mechanistic precision that is transforming both biological discovery and the development of next-generation spatial biomarkers for patient stratification and therapeutic targeting.
The Common Thread
Each generation directly addressed the blind spot of the one before it. Bulk RNA-seq replaced noisy microarray averages with digital transcriptome-wide quantification. scRNA-seq replaced tissue-level averages with single-cell resolution. Spatial transcriptomics restored the spatial context that dissociation-based methods destroyed. Together, the three platforms are not competing technologies but a layered toolkit — each occupying a distinct and complementary niche — and the convergence of spatial transcriptomics with modern AI represents the current frontier of what is scientifically and clinically achievable from a tissue biopsy.